Pairwise boundary and IAS-proxy method selection
An exhaustive numerical study considered returning every local minimum of the neutral-promolecular line density between two atoms. Although this contract is possible, it makes interpolation-scale features, one-ULP knot behavior, sub-ULP event ordering, and exact tie handling part of the public science.
That is more precision than the intended geometry workflow needs. atomref
therefore exposes two explicit methods instead of hiding one policy behind a
single number.
Recommended default: proatomic boundary
The default method returns a stable neutral-proatom divider:
- identical atoms use the exact midpoint;
- overlapping unlike atoms use the point where the two neutral proatomic densities are equal;
- separated low-density contours use the midpoint of the contour gap;
- complete one-atom dominance is reported explicitly.
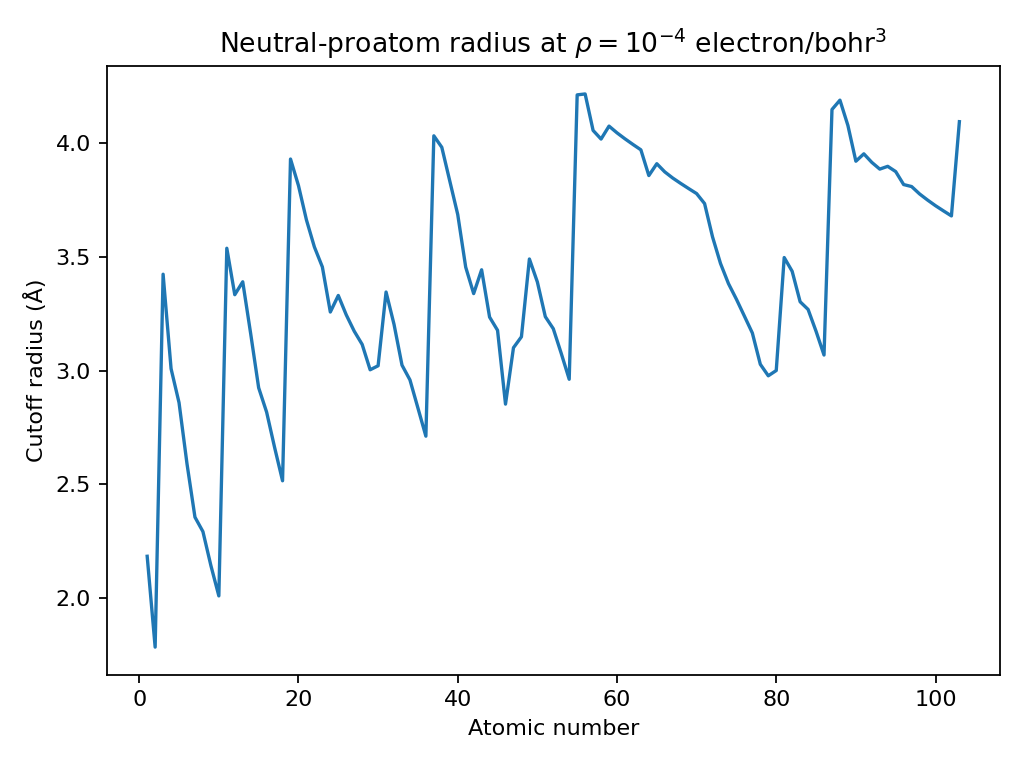
The fixed per-atom tail cutoff is
1e-4 electron/bohr^3. It is a model policy for ignoring weak neutral-proatom
tails, not a universal QTAIM interaction threshold.
Optional: practical promolecular minimum
The optional minimum method is retained for Bader-oriented comparison and calibration. It searches the sum of the two proatomic densities only where both components remain above the cutoff.
The search has a declared spatial resolution of 0.01 bohr. It returns one
resolved minimum and, when useful, one competitive alternative. It does not
attempt to preserve every mathematical microminimum. Raw refined candidates
from the required 0.02 and 0.01 bohr passes, and from the 0.005 bohr
fallback when used, are combined before one resolution-coalescing step.
Position-sorted candidates connected by successive gaps below 0.01 bohr
represent one resolved valley. Distinct adjacent binary64 grid coordinates are
retained so both endpoints and the midpoint remain available near cutoff
contact. Refined cutoff endpoints and nuclei are discarded rather than clamped.
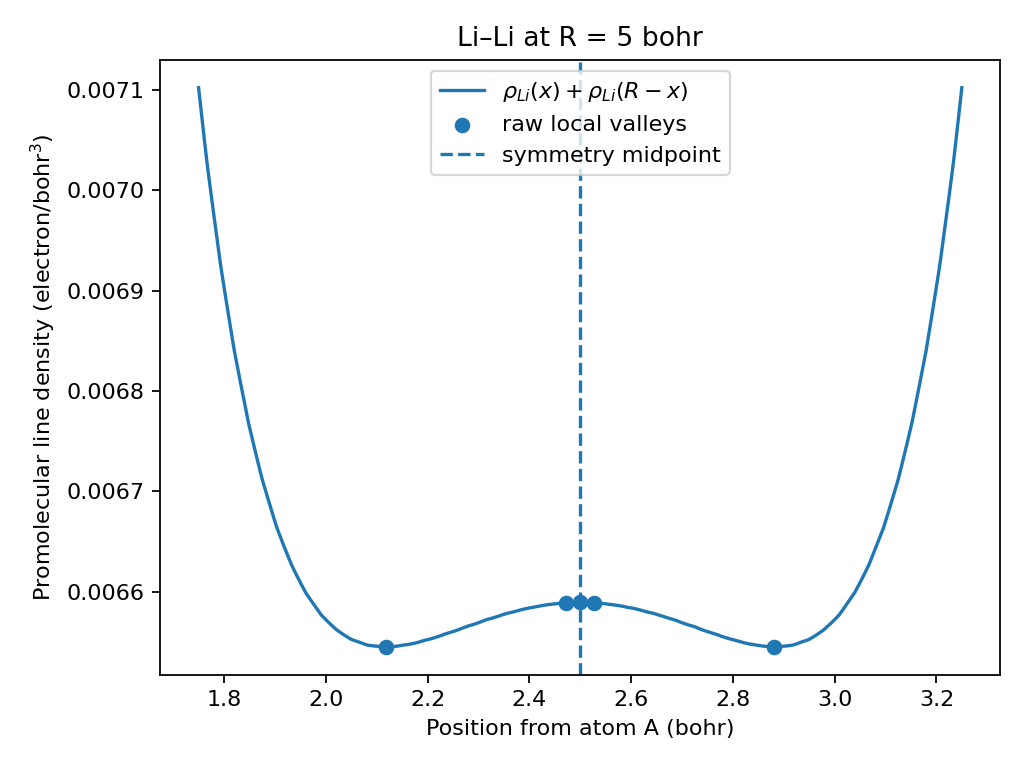
For identical atoms, symmetry still fixes the returned coordinate at R/2.
The Li–Li example shows why this rule is necessary: the raw interpolant has
slightly deeper symmetric off-centre valleys, but either one alone is a poor
separator for identical atoms.
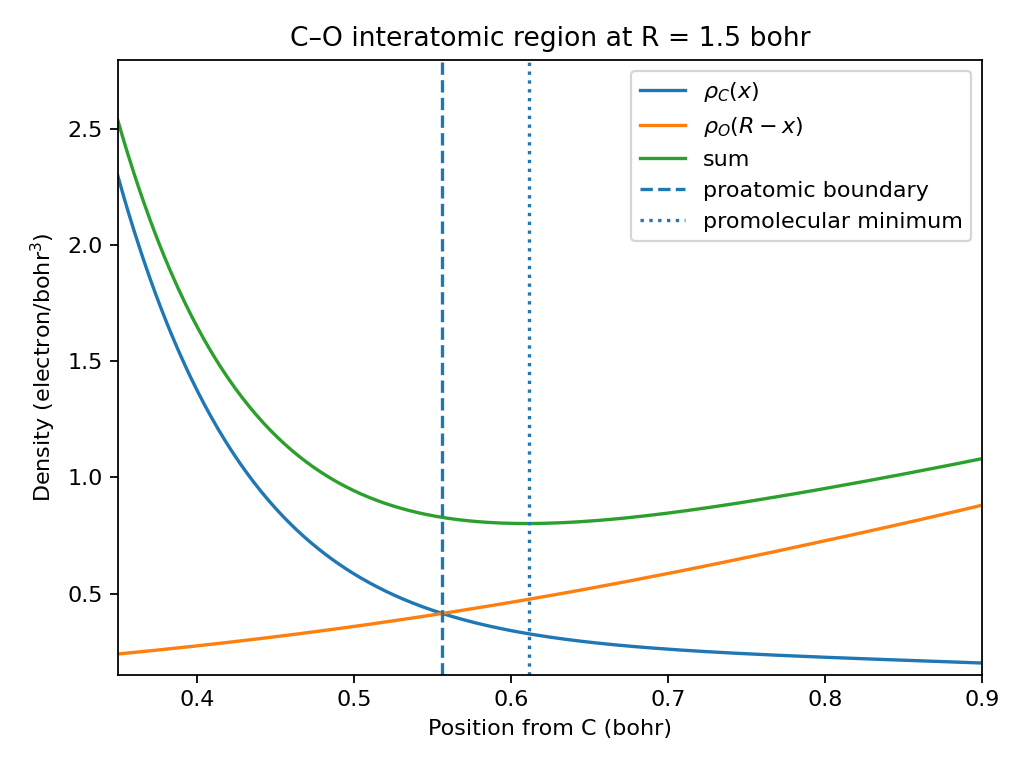
The two methods are scientifically different. For C–O at 1.5 bohr, the balanced-contribution coordinate and the promolecular minimum do not coincide. That disagreement is expected rather than a solver defect.
Numerical evidence
The executed study compared the practical minimum search with a slower
0.001 bohr reference over 300 deterministic H–Lr cases and seven adversarial
cases selected from the exhaustive all-minima analysis.
- All 298 cases where both methods returned a minimum agreed within
0.01 bohr. - The largest coordinate difference was about
0.00129 bohr. - Two extremely short-distance reference minima were intentionally reported as unresolved because they were narrower than the public practical resolution and lay immediately beside a nucleus.
- On the machine used for the study, the standalone implementation took roughly
0.05–0.06 msper cached boundary estimate and about0.8 msper cached practical minimum estimate.
These figures are evidence, not portable performance guarantees.
Why exhaustive all-minima search is not the default
The exhaustive investigation found real edge cases:
- a minimum only 27 binary64 coordinates from a profile knot;
- one-ULP inversions at exact knots;
- multiple shallow or symmetric minima, including five for Li–Li at 5 bohr;
- fourteen H–U minima in a case where a boundary value was lower;
- distinct exact events that rounded to one float;
- value errors large enough to complicate exact global-tie classification.
Those facts matter under an “expose every local minimum” contract. They do not materially improve a stable pairwise divider, and the practical minimum mode coalesces or rejects structure below its declared resolution.
Public API
estimate_proatomic_boundary(...)
estimate_promolecular_density_minimum(...)
estimate_ias_position(..., mode="boundary" | "minimum")
boundary is the default. The dispatcher never silently switches modes.
Neither result is an exact molecular-density QTAIM surface.
Scientific context
The interpretation follows three established ideas while keeping their scopes separate:
- QTAIM basin boundaries are zero-flux surfaces of the molecular electron density: Bader, Chemical Reviews 91 (1991), 893–928, doi:10.1021/cr00005a013.
- Hirshfeld stockholder weights are proportional to free-atom reference densities, which motivates equal pairwise contributions as a stable divider: Hirshfeld, Theoretica Chimica Acta 44 (1977), 129–138, doi:10.1007/BF00549096.
- Radial-density atom and bond constructions are useful model definitions but require comparison with molecular-density results: Warburton, Poirier, and Nippard, J. Phys. Chem. A 115 (2011), 852–867, doi:10.1021/jp1093417.
The complete calculations, representative profiles, adversarial cases, and
local timing record are in the directly rendered
04-ias-method-selection-study.ipynb
notebook. Its committed outputs are shown without re-execution during the
documentation build.